METLIN
Use of METLIN in Metabolomics
Imagine after completing your metabolomics experiment you get results where you get a long list of mysterious chemical signals from a mass spectrometer. Your big question is: “Which molecules are these?”
That’s exactly the problem METLIN was created to solve. In the early days of metabolomics, scientists struggled to identify metabolites quickly and confidently. Without a reliable reference library, they had to compare their data against scattered, small-scale, or theoretically predicted spectra—a slow and often inaccurate process. METLIN was built to act like a “chemical fingerprint database.” Researchers at The Scripps Research Institute experimentally analyzed thousands of known metabolite standards using tandem mass spectrometry (MS/MS) and stored their unique spectral “fingerprints” in METLIN. Now, when scientists run their own samples, they can search their unknown signals against METLIN’s curated library to get fast, reliable matches.
E.g. you’re have to compare the blood samples from healthy individuals and patients with a specific disease. Your instrument detects a signal that’s much stronger in the patient group.
Without METLIN: You’d know something is different, but figuring out what it is could take weeks of extra experiments and guesswork.
With METLIN: You immediately search that signal’s MS/MS “fingerprint” against the database. Within minutes, you get a match—let’s say it’s lactate. Now you don’t just know a “peak is higher”; you know lactate levels are elevated, which could point to disrupted energy metabolism in the disease. This turns a vague observation into a clear biological insight.
In short: METLIN turns unknown peaks into identified molecules, making metabolomics more reproducible, high-throughput, and discovery-friendly. It’s like a universal reference book of metabolite identities that the whole research community can use.
So What is METLIN?
The METLIN Metabolite and Chemical Entity Database is a large, publicly accessible scientific database used in metabolomics research. It contains detailed information on metabolites, small molecules, and chemical compounds found in biological systems. METLIN helps researchers identify unknown metabolites by comparing experimental mass spectrometry data with reference data stored in the database. The database includes accurate mass values, molecular structures, chemical formulas, and fragmentation patterns. It is widely used in biomedical research, drug discovery, toxicology, nutrition, and environmental studies. In simple terms, METLIN acts like a reference library that helps scientists recognize and understand small molecules in biological samples. Metabolomics tells us that a molecule is changing; METLIN helps us identify what that molecule is.
METLIN is directly related to metabolomics because it supports the identification and interpretation of metabolites.
Here is the Link to explore METLIN
How to use/navigate METLIN?
Methodology to use METLIN
Case scenario/research question
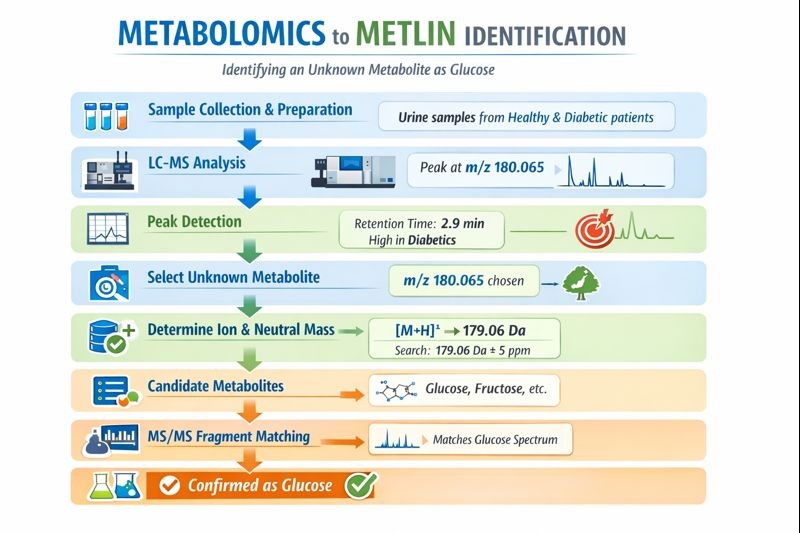
A metabolomics study compares urine samples from healthy individuals and diabetic patients. One metabolite peak is significantly higher in diabetic samples and needs to be identified.
Steps to navigate METLIN
Step 1: Sample preparation
Collect the Urine samples from both groups, centrifuge and filter to remove proteins and impurities while preserving small metabolites and get clear metabolite extracts
Step 2: LC–MS analysis
Analyze the prepared samples using liquid chromatography–mass spectrometry to separate metabolites and measure their mass-to-charge ratios (m/z). LC–MS analysis produces hundreds of peaks. One peak is observed to appear at m/z 180.065.
Step 3: Peak detection
Data-processing software detects peaks and records their m/z value, retention time, and intensity. e.g. the peak is at m/z 180.065, retention time is 2.9 minutes , and it shows much higher intensity in diabetic samples.
Step 4: Selection of an unknown metabolite
A peak of interest is selected based on biological relevance or statistical significance. The researcher can select the m/z 180.065 peak because it clearly differentiates healthy and diabetic groups.
Step 5: Determine ion mode and adduct
The ionization mode* and ion form* are identified to calculate the neutral mass*. So e.g. the peak is detected in positive ion mode as [M+H]+ , indicating a neutral mass of approximately 179.06 Da. Meaning of [M+H]⁺
- M = the original (neutral) molecule
- +H = one proton (H⁺) is added
- ⁺ = overall positive charge
So [M+H]⁺ means: The molecule gained one proton and is detected as a positively charged ion.
Side notes: Let us try to understand what is ionization mode; ion; neutral mass and it’s significance in METLIN
What does “ionization mode” mean?
Mass spectrometers cannot detect neutral (uncharged) molecules. They can only detect ions (charged molecules). To make molecules detectable, they are ionized during analysis. There are two common ionization modes:
- Positive ion mode → the molecule gains a positive charge
- Negative ion mode → the molecule gains a negative charge
What does “ion form” mean?
When a molecule becomes charged, it usually adds or loses a small ion. The way the molecule appears in the mass spectrometer is called the ion form (or adduct) , such as:
- H⁺ (proton)
- Na⁺ (sodium)
- K⁺ (potassium)
- H⁻ (hydride loss)
What is Neutral mass?
Neutral mass is the actual molecular weight of a compound when it has no electrical charge. So it is the true mass of the molecule itself, before it gains or loses any ions during mass spectrometry. It is like a person’s body weight without accessories
Why “neutral mass” is needed?
Mass spectrometers do not measure neutral molecules. They detect charged forms (ions) of molecules.
During analysis, a molecule may:
gain a proton (H⁺) → positive ion
lose a proton (H⁺) → negative ion
bind sodium or potassium ions
Because of this, the mass seen by the instrument is not the molecule’s true mass. Hence to identify the compound correctly, scientists calculate the neutral mass and use that value to search databases such as METLIN. E.g.
Detected ion: [M+H]⁺
Measured m/z: 180.065
Mass of H⁺: ≈ 1.007 Da
Neutral mass
= 180.065 − 1.007
≈ 179.06 Da
This value (179.06 Da) is the neutral mass of the molecule.
Let us say that the ionized mass is the person wearing a backpack then the neutral mass is the person alone. So to identify the person correctly, you need the weight without the backpack.
Step 6: Search METLIN database
The accurate mass is entered into METLIN with an appropriate mass tolerance. E.g. you can search METLIN for 179.06 Da ± 5 ppm in positive mode.
Step 7: Review candidate metabolites
METLIN will provide a list of metabolites with matching masses. METLIN may suggest several candidates, including glucose, fructose, and other sugar-related metabolites.
Step 8: MS/MS spectrum comparison
The fragmentation pattern of the unknown metabolite is compared with reference spectra in METLIN. E.g. the MS/MS fragments of the unknown peak may closely match with the glucose fragmentation pattern in METLIN.
Step 9: Biological and chromatographic validation
Retention time behavior and biological relevance are evaluated. Glucose is biologically relevant to diabetes and elutes at a retention time consistent with polar sugars.
Step 10: Confirmation and reporting
If available, an authentic standard is used to confirm identity. A glucose standard can be run and if shows the same retention time and MS/MS spectrum, that confirms the metabolite as glucose.
It is important to remember that Mass match alone is not enough because many compounds can share similar/identical masses (isomers). A strong identification uses accurate mass + MS/MS match + retention time + (ideally) authentic standard.